- GUI for Quantum ESPRESSO
- Manual download Quantum ESPRESSO binaries
- Quantum ESPRESSO
- Quantum ESPRESSO
- Plane-wave periodic DFT with GUI
- Plane waves vs. atomic-orbital based periodic DFT
- Download
- Quantum espresso для windows
- BragitOff.com
- READ-LEARN-BRAG!
- Installing Quantum Espresso on Windows – TUTORIAL
- DOWNLOAD:
- INSTALLATION:
GUI for Quantum ESPRESSO
Starting with our 2017 release, you can use the integrated graphical interface also to set up and visualize Quantum ESPRESSO plane wave calculations. The GUI will prompt you to download the binaries and pseudopotentials automatically when needed. In the Amsterdam Modeling Suite 2018 we ship Quantum ESPRESSO 6.3 binaries.
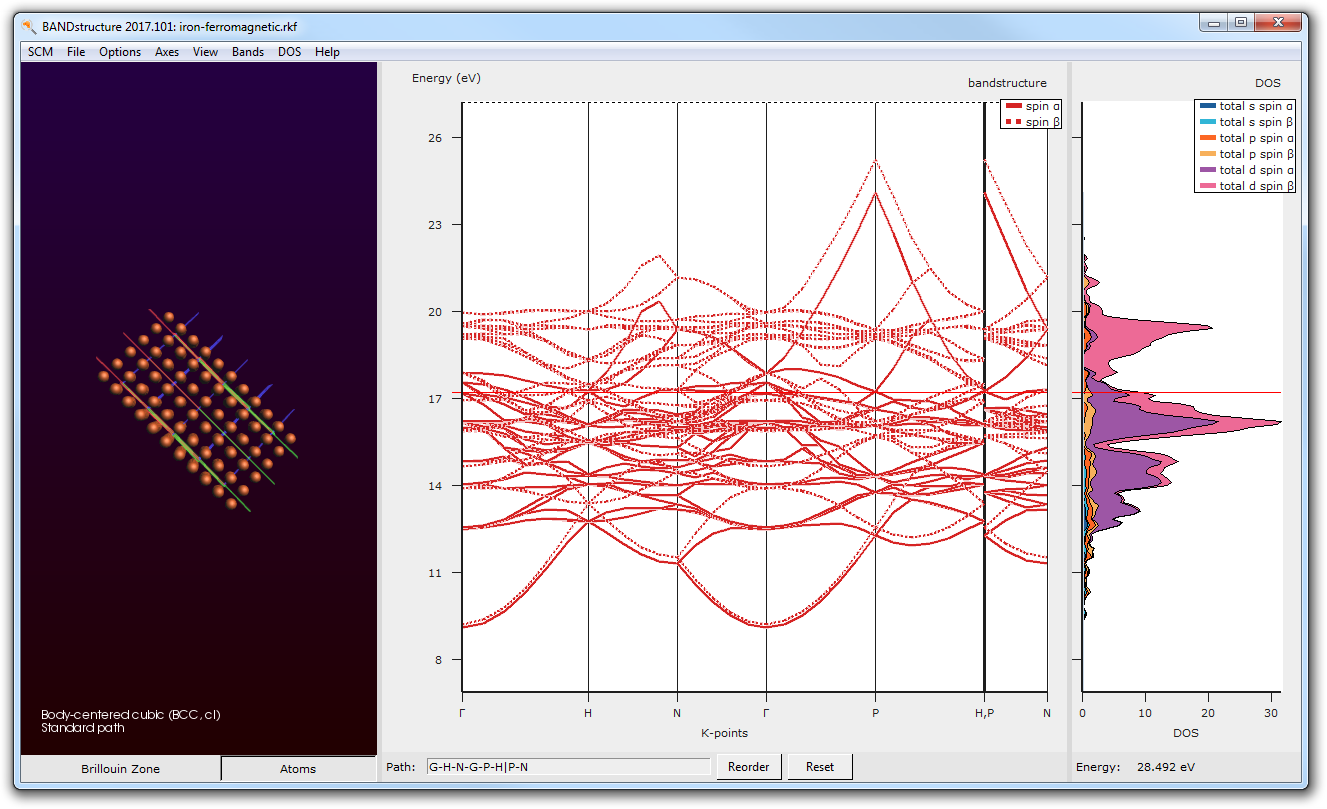
The current interface allows you to set up geometry and lattice optimizations, as well as visualizing electronic properties such as (partial) DOS, band structures and spin polarization. Just request a free trial and go through the Quantum ESPRESSO GUI tutorials to see how easy it is.
Manual download Quantum ESPRESSO binaries
If the automatic download from the GUI doesn’t work, e.g. because of firewall issues, you can also download the Windows, Mac OS and Linux Quantum ESPRESSO 6.3 binaries separately. Similarly you can also download the pseudopotential library separately, which include the Quantum ESPRESSO pseudopotentials (6.2.1) and the pseudopotentials from Garrity, Bennet, Rabe and Vanderbilt (GBRV).
If you don’t change the GUI environment variables, install the binaries in $ADFBIN and the pseudopotential in $ADFHOME/atomicdata on Windows or Linux. On a Mac both go in «$HOME/Library/Application Support/SCM» . The binaries should be in a folder qe6.3 , and the pseudo potentials in a folder upf_files .
Please cite Quantum ESPRESSO according to the reference as well as any underlying functionality (functionals, pseudopotentials).
Quantum ESPRESSO
Fast plane wave periodic DFT + great GUI
Build, optimize, band structures, DOS
Out of the box binaries Windows, Mac, Linux
Quantum ESPRESSO
Plane-wave periodic DFT with GUI
Quantum ESPRESSO is an open-source plane-wave periodic density functional theory code, the active development of which is coordinated by the Quantum ESPRESSO foundation. The SCM team has built binaries for single-node Windows, Mac & Linux, which you can download & use straight from the Graphical User Interface. With our 2019 release of the Amsterdam Modeling Suite (AMS2019) we ship Quantum ESPRESSO 6.3 binaries for Windows, MacOS & Linux.
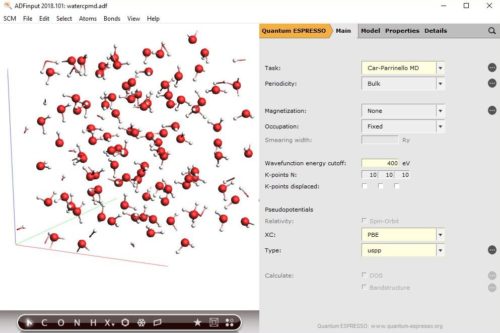
The GUI makes getting started with Quantum ESPRESSO a breeze: it will download and install the binaries and pseudopotentials for you. Furthermore the GUI supports building structures, running & visualizing geometry optimizations, band structures, and (partial) DOS. There is also support for Car-Parrinello MD (CPMD).
Future projects include phonons, electron-phonon coupling and other properties. Let us know which features are of most interest to you!
Within the Amsterdam Modeling Suite it is also easy to switch between Quantum ESPRESSO and any of our other modules, such as the atomic orbital-based periodic DFT code BAND.
Plane waves vs. atomic-orbital based periodic DFT
Plane-wave periodic codes such as Quantum ESPRESSO are usually fast for dense many-electron systems by virtue of the pseudopotential or projector-augmented wave approximation. This is why SCM started developing GUI support for it, while still continuing the development of our own AO-based periodic DFT code BAND.
The atom-centered basis functions ansatz in BAND offers a number of interesting features which complement speedy plane-wave calculations:
- Compare clusters with slabs using the same basis set
- Easy analysis of orbital & densities (pDOS, fat bands, COOP, MOs, …)
- No pseudopotential approximations – basis sets for all elements, all-electrons
- Core electron properties: EPR, core holes, …
- True 1D and 2D periodicity (surface polarization, electric fields, …)
- Modern xc functionals: SCAN, MN15-L, HSE06, GLLB-sc, D3(BJ), …
- Self-consistent NEGF, including gate & bias potential, spin transport
- Fully self-consistent spin-orbit coupling (SCF, not perturbative as in plane-wave codes)
Try the Amsterdam Modeling Suite yourself!
Download
How to cite?
Quantum ESPRESSO is an Open Source distribution. We shall greatly appreciate if scientific work done using Quantum ESPRESSO will contain an explicit acknowledgement and the following references:
and if you used the GPU-enabled version of the code:
P. Giannozzi et al., J. Chem. Phys. 152, 154105 (2020) https://doi.org/10.1063/5.0005082
How to download?
Sources: Quantum ESPRESSO is currently distributed as source packages, but selected binary packages for Linux, Mac-OS X and Windows are also available. The current stable version can be downloaded from:
- GitHub (recommended), or alternatively from
- GitLab (click on the «cloud with a down arrow» to download);
- GitLab repository of GPU-enabled version.
Please read the «release-notes» file for information on major changes and problems. Patches and updates are available on GitHub: see the «Assets» list at the end of the release notes. The latest development version, in branch «develop», is available on GitLab and mirrored on GitHub.
The Schrödinger-enabled version of Quantum ESPRESSO can be downloaded here.
The RISM-enabled version of Quantum ESPRESSO can be downloaded from Satomichi Nishihara’s git repository.
Virtual Machine: A fully configured Ubuntu virtual machine that can be run from Windows/Mac-OS x/Linux/Solaris is available on the Materials Cloud site. It contains Quantum ESPRESSO and much more (4Gb).
Binaries: Binary packages for Linux are available in the Debian stable and unstable distributions, courtesy Michael Banck and DebiChem team.
Binary packages for Mac-OS X are available in the Science section of Macports.
Windows binaries are made available by AdvanceSoftware Corp.. See also the SCM site.
Quantum espresso для windows
pietrodelugas released this Dec 2, 2020
New in 6.7 version:
- Support for CMake (F. Ficarelli and D. Cesarini, CINECA, with help from
Ye Luo, P. Delugas, S. Gsaenger) - In vc-relax with Hubbard corrections, the final SCF calculation is done by
reading atomic occupations from file produced during the vc-relax
(rather then recomputing them from scratch). - EPW:
(1) ZG package to generate special displacements for first-principles non-perturbative calculations
at finite temperatures [Marios Zacharias and Feliciano Giustino, Phys. Rev. Research 2, 013357, (2020)].
(2) Plotting of Fermi surface.
For the full list of new features, bug fixes, and changes leading to backward incompatibility issues,
please visit the Releases page of the EPW documentation site [https://docs.epw-code.org/doc/Releases.html].
Fixed in 6.7 version:
- Some linkers yield «missing references to ddot_» in libbeef
- FFT test in FFTXlib was not always compiling
- angle1, angle2, starting_magnetization incorrectly written to xml file
- Bug in Hubbard forces and stress for bands parallelization (when nproc_pool>nbnd)
- Bug in DFT+U+V when starting_ns_eigenvalue is used (courtesy of M. Cococcioni)
- Crash in the calculation of Z* with ultrasoft PP when the number of bands
is larger than the number of occupied bands (thanks to Sasha Fonari) - Crash in matdyn.x when ibrav=0 (thanks to Sasha Fonari)
- Some postprocessing cases not working properly with k-point parallelization
- Ensemble dynamics in CP was broken in v.6.6 (not in previous versions)
Incompatible changes in 6.7 version:
- FoX no longer used to read and write pseudopotential files
- iotk no longer used to read and write any file
- Developer manual moved to the Wiki
BragitOff.com
READ-LEARN-BRAG!

Installing Quantum Espresso on Windows – TUTORIAL


Quantum Espresso is an integrated suite of Open-Source computer codes for electronic-structure calculations and materials modelling at the nanoscale. It is based on density-functional theory, plane waves, and pseudopotentials.
I have already shown you how to download, install and run Quantum Espresso on Linux here.
In this post I will do the same for Windows.
DOWNLOAD:
In my limited knowledge I noticed that the latest version of Quantum Espresso, that is version 6.1, isn’t available for windows. As one can check here: http://qe-forge.org/gf/project/q-e/frs/?action=FrsReleaseBrowse&frs_package_id=18
The latest version available for Windows is 5.3.
You can download it from here.
The binaries available for both 32 and 64-bit versions. You can download them according to your pc configurations.
It should also be noted that QE on windows uses mpich for parallel processing.
INSTALLATION:
Once downloaded, the installation procedure is really simple. Just double click on the installer(setup) and follow the instructions.
If you successfully installed QE then, you can run a simple demo now to see if the things work as they should.
Let’s perform an SCF calculation for Silicon for demo purpose.
To perform such a calculation, you need an input file for Quantum Espresso. You can download the sample input file from this link(si.scf). Once you have the file, save it in a folder of your choice. For the sake of this tutorial, let’s say you save it in a folder called ‘QE’ inside the Quantum Espresso installation directory.
Now, create a folder called ‘temp’ inside the directory that contains the input file(in our example the folder is ‘QE’).
Now let me tell you a little about the input file that we have here. In this file, we are basically trying to find out some of the properties for a Si atom, like the total energy, etc.
The input file is a really important file and you will need to learn quite a few things to be able to build one yourself.
There are a lot of things that need explaining there, but I guess I will leave that for another post, and in this tutorial we will, just focus on running our first pw scf calculation. For the sake of the tutorial you can download the input file that I will be using from here(Si_Quantum_Espresso_input) or you can just copy the following and save it in a file called si.scf.in .
&CONTROL
calculation = «scf»
max_seconds = 8.64000e+04
pseudo_dir = «F:/QE/»
outdir=»temp»
/
&SYSTEM
a = 5.46873e+00
degauss = 1.00000e-02
ecutrho = 1.00000e+02
ecutwfc = 2.50000e+01
ibrav = 1
nat = 8
ntyp = 1
occupations = «smearing»
smearing = «gaussian»
/
&ELECTRONS
conv_thr = 1.00000e-06
electron_maxstep = 200
mixing_beta = 7.00000e-01
startingpot = «atomic»
startingwfc = «atomic+random»
/