- Quantum espresso windows установка
- Download
- 1 Installation
- 1.1 What Fortran compiler do I need?
- 1.2 Why is configure saying that I have no fortran compiler?
- 1.3 Why is configure saying that my fortran compiler doesn’t work?
- 1.4 configure doesn’t recognize my system, what should I do?
- 1.5 Why doesn’t configure recognize that I have a parallel machine?
- 1.6 Compilation fails with _internal error_, what should I do?
- 1.7 Compilation fails at linking stage: _symbol … not found_
- 1.8 Compilation works but the executable doesn’t start because _shared libraries … not found_
- Easiest Way to Install Quantum Espresso on Windows
- How to install quantum espresso on windows
Quantum espresso windows установка
Installing the Quantum ESPRESSO distribution
How to get the distribution, how to install it, i.e. produce the executables; what you need for a succesful installation, and what to do if the installation is NOT successful! Updated September 2019
Requirements for Quantum ESPRESSO installation
Things that you MUST have on your machine:
- Unix, or a Unix-like environment (a shell and the Make utility): Mac OS-X ok, Windows with «cygwin» ok
- a working fortran compiler compliant with the F2003 standard (not-so-old compilers, e.g., «gfortran», should do the job)
- a working C compiler («gcc» is ok)
All hardware is supported, as long as it has what is listed above, BUT: in order to exploit GPU’s, you need to compile the GPU-enabled version of QE. In order to run in parallel, you MUST have at least one of the following:
- working MPI (Message-passing Interface): parallel compiler («mpif90» or similar scripts), MPI libraries, run-time environment («mpirun» or similar launchers)
- OpenMP-capable compiler and autothreading mathematical libraries (for multi-core CPUs)
Libraries: Quantum ESPRESSO uses and provides a copy of the following external libraries:
- BLAS (Basic Linear Algebra Subroutines): http://www.netlib.org/blas
- LAPACK (Linear Algebra Package): http://www.netlib.org/lapack
- FFTW (Fast Fourier-Transform package): http://www.fftw.org
Things that you SHOULD have on your machine for real-life usage:
- Fast mathematical libraries
- For parallel execution: fast interprocess communication hardware and software
- Choose and create a directory where to install Quantum ESPRESSO. It should be on a file system that
- is local to the PC you are using: sometimes the home directory in a PC cluster is accessed via the network (NFS). Moving large amount of data via the network MUST BE AVOIDED.
- has enough disk space and a large enough disk quota: sometimes the home directory is small, or has a quota enforced. The compiled complete distribution may take in the order of 1 Gb.
In this tutorial, Quantum ESPRESSO is installed in directory «/afs/ictp/public/shared/smr2522/»
- Download in the chosen directory the package qe-XYZ.tar.gz (XYZ=version number).
The suffix «.gz» means «compressed by gzip» (a free utility found on most Unix machines).
The suffix «.tar» means «archived by tar» (the standard Unix command for archiving and retrieving files) In this tutorial, Quantum ESPRESSO has already been downloaded . - Uncompress and unpackage the file: On some machines the «-» is not needed, or the «z» flag (meaning «uncompress files compressed by gzip») is not supported. If so: A directory «qe-XYZ/» will be created, containing many files and other directories. In this tutorial, Quantum ESPRESSO has already been unpacked .
- Execute initialization steps, if needed. For instance, in order to enable the usage of Intel compiler and MKL libraries, you should set a number of variables; for parallel execution, you should have a parallel compiler in your path. How to do this depends on the specific machine. Sometimes one has to use the command «module», as in the following example: In this tutorial, command «source /opt/intel/2011/composerxe/bin/compilervars.csh intel64» has been executed before compilation .
- Enter the «qe-XYZ/» directory and execute «./configure»: «configure» is a wrapper, calling the «install/configure» script, that tries to guess your machine and to choose compilation and linking options accordingly («install/configure» is generated from «install/configure.ac» using the complex but well-known Unix utility «autoconf»). The result of «configure» is a file called «make.inc» containing the compilation and linking options. You may want to have a look at it to verify what «configure» thinks about your system. Several options to «configure» may (and sometimes, have to) be specified (see below).
- If you get a message «architecture xxx not recognized», specify option «ARCH=. «, such as e.g. (for a BlueGene machine):
- If «configure» selects a compiler you don’t like (or one that doesn’t like Quantum ESPRESSO) specify option «MPIF90=. » (also for serial case), e.g.:
If everything is fine you should get a bunch of (mostly obscure and irrelevant) messages but no error. Read the last lines: you may need to understand them if something goes wrong at compilation stage, or if you need to boost performances.
NOTE: if you have a parallel compilation script like «mpif90» in you path, «configure» will choose it. With OpenMPI, you can use environment variable OMPI_FC to select the compiler used by the «mpif90» script. Use to produce a serial executable. Use to produce a parallel executable with OpenMP enabled. In this tutorial, we used «./configure —disable-parallel CC=gcc» for serial compilation; for parallel compilation, we need command «setenv OMPI_MPIFC ifort», then «./configure CC=gcc» - Compile the packages you need. Let us start from PWscf: ([] is optional: «-j N» executes «make» in parallel on N processors to speed up compilation) «make» is another complex but standard unix utility that compiles what is needed in the proper order (for instance: in Fortran, you need to compile modules before programs that use them). The configuration files (Makefiles, dependencies) for «make» are contained n the distribution. If everything goes well, executables will appear in «bin/»:
- Not-so-quick test (for «pw.x» executable only) to verify that things look good: on a parallel machine, or, on a serial one,
- Compile other packages. «make» with no argument yields a list of targets, i.e., packages to be compiled. If you want to compile everything: Some additional packages (notably, W90) that are not contained in the QE tarball will be download from the net. Beware: this will work only if you have direct access to the internet, and working «wget» or «curl» commands. If not, you will need to download the required packages into the «archive/» directory. Then you can do «make» again. In this tutorial, everything is already installed .
Requirements for installation of a FAST executable
Most of the CPU time in a typical run is spent in:
- Fast Fourier Transform (FFT)
- matrix-matrix and matrix-vector multiplications (BLAS)
- solution of linear systems, diagonalizations (LAPACK)
If you want a fast executable, you need machine-optimized BLAS, LAPACK, FFT libraries, and in parallel execution, ScaLAPACK (if possible, ELPA).
Download
How to cite?
Quantum ESPRESSO is an Open Source distribution. We shall greatly appreciate if scientific work done using Quantum ESPRESSO will contain an explicit acknowledgement and the following references:
and if you used the GPU-enabled version of the code:
P. Giannozzi et al., J. Chem. Phys. 152, 154105 (2020) https://doi.org/10.1063/5.0005082
How to download?
Sources: Quantum ESPRESSO is currently distributed as source packages, but selected binary packages for Linux, Mac-OS X and Windows are also available. The current stable version can be downloaded from:
- GitHub (recommended), or alternatively from
- GitLab (click on the «cloud with a down arrow» to download);
- GitLab repository of GPU-enabled version.
Please read the «release-notes» file for information on major changes and problems. Patches and updates are available on GitHub: see the «Assets» list at the end of the release notes. The latest development version, in branch «develop», is available on GitLab and mirrored on GitHub.
The Schrödinger-enabled version of Quantum ESPRESSO can be downloaded here.
The RISM-enabled version of Quantum ESPRESSO can be downloaded from Satomichi Nishihara’s git repository.
Virtual Machine: A fully configured Ubuntu virtual machine that can be run from Windows/Mac-OS x/Linux/Solaris is available on the Materials Cloud site. It contains Quantum ESPRESSO and much more (4Gb).
Binaries: Binary packages for Linux are available in the Debian stable and unstable distributions, courtesy Michael Banck and DebiChem team.
Binary packages for Mac-OS X are available in the Science section of Macports.
Windows binaries are made available by AdvanceSoftware Corp.. See also the SCM site.
1 Installation
Most installation problems have obvious origins and can be solved by reading error messages and acting accordingly. Sometimes the reason for a failure is less obvious. In such a case, you should look into Sec.2, Installation, of the user guide, and into the archive of the users’ mailing list to see if a similar problem (with solution) is described. If you get really weird error messages during installation, look for them with your preferred Internet search engine (such as Google): very often you will find an explanation and a workaround.
Also please have a look at the following page by Glenn Lockwood, containing a lot of useful info.
1.1 What Fortran compiler do I need?
Any non-buggy, or not-too-buggy, fortran-95 compiler should work, with minimal or no changes to the code. configure may not be able to recognize your system, though.
1.2 Why is configure saying that I have no fortran compiler?
Because you haven’t one (really!); or maybe you have one, but it is not in your execution path; or maybe it has been given an unusual name by your system manager. Install a compiler if you have none; if you have one, fix your execution path, or define an alias if it has a strange name. Do not pass an executable with the path as an argument to configure, as in e.g. ./configure F90=/some/strange/f95: it doesn’t work.
1.3 Why is configure saying that my fortran compiler doesn’t work?
Because it doesn’t work (really!); more exactly, configure has tried to compile a small test program and hasn’t succeeded. Your compiler may not be properly installed. For Intel compiler on PC’s: you may have forgotten to run the required initialization script for the compiler.
1.4 configure doesn’t recognize my system, what should I do?
If compilation/linking works, never mind; otherwise, try to supply a suitable supported architecture, or/and manually edit the make.sys file. Detailed instructions in Sec.2 of the user guide.
1.5 Why doesn’t configure recognize that I have a parallel machine?
You need a properly configured complete parallel environment. If any piece is missing, configure will revert to serial compilation. Detailed instructions in Sec.2 of the user guide.
1.6 Compilation fails with _internal error_, what should I do?
Any message during compilation saying something like internal compiler error and the like means that your compiler is buggy. You should report the problem to the compiler maker – especially if you paid real money for it. Sometimes reducing the optimization level, or rearranging the code in a strategic place, will make the problem disappear. In other cases you will need to move to a different compiler, or to a less buggy version (or buggy in a different way that doesn’t bug you) of the same compiler.
1.7 Compilation fails at linking stage: _symbol … not found_
If the missing symbols (i.e. routines that are called but not found) are in the code itself: most likely the fortran-to-C conventions used in file include/c_defs.h are not appropriate. Edit this file and retry.
If the missing symbols are in external libraries (BLAS, LAPACK, FFT, MPI libraries): there is a name mismatch between what the compiler expects and what the library provides. See Sec.2 of the user guide.
If the missing symbols aren’t found anywhere either in the code or in the libraries: they are system library symbols. i) If they are called by external libraries, you need to add a missing system library, or to use a different set of external libraries, compiled with the same compiler you are using. ii) If you are using no external libraries and still getting missing symbols, your compiler and compiler libraries are not correctly installed.
1.8 Compilation works but the executable doesn’t start because _shared libraries … not found_
This is a frequent problem with MKL libraries. You need to set some environment variables telling the system where the shared libraries are. See the documentation by Intel that comes with MKL libraries.
Easiest Way to Install Quantum Espresso on Windows
I was looking for a way to install quantum espresso on my windows PC on search engine but all the information I needed wasn’t in a particular page. I download some videos on how to install quantum espresso, read some articles online, and download articles in PDF before I can be able to install it.
Therefore I have decided to put into this page what I did that enabled me to install quantum espresso and run my first program on it successfully.
The steps are simple and will be accompanied with images for better understanding.
How to install quantum espresso on windows
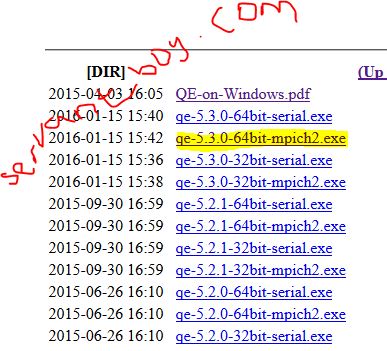
- Visit this website to download espresso http://rpm.lammps.org/qe4win/release/index.html. In my case I downloaded qe-5.3.0-64bit-mpich2.exe. The file is for 64-bit windows (that of 32-bit windows is there also)
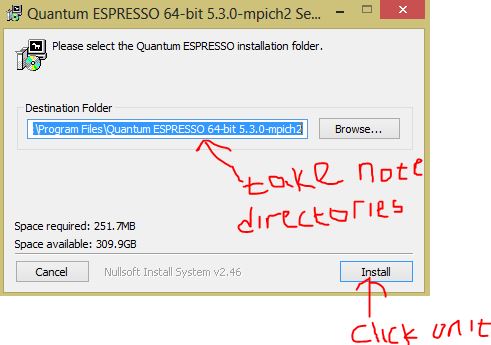
- Click on the downloaded the file to install it (Please take note of the destination of the folder (directory))

- After you have finished installing, then
- Visit this website to download mpich2 (www.mpich.org/static/tarballs/1.4.1p1/mpich2-1.4.1p1-win-x86-64.msi). And install it on your windows PC. The file is for 64-bit windows (that of 32-bit is there also).
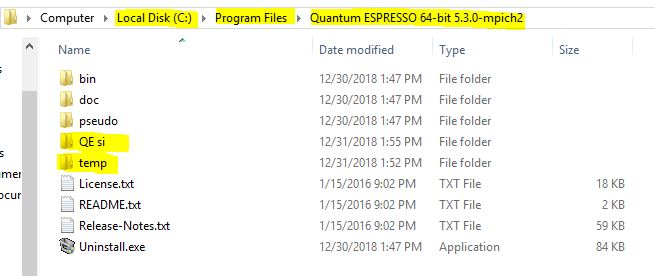
- The next thing is to locate the folder destination of the installed quantum espresso on your windows PC. In my own case it was found in Local disk (c:) (program file).
Next is to perform your first simple calculation to know whether you have successfully installed quantum espresso on your windows PC
- Create an input file and put it inside a folder name QE si. The input file is for silicon
- Create a folder and name it temp and put it inside the destination folder of quantum espresso
- Open the input file and put your pseudo directory inside the double colum pseudo_dir = ”” and also put your temp directory inside the double column of the outdir = “”
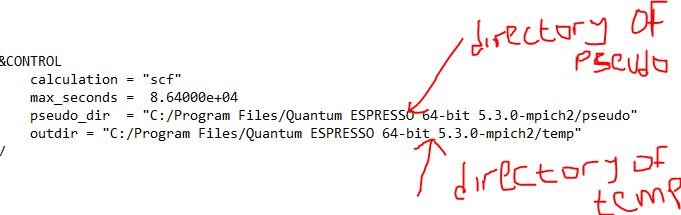
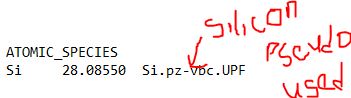
- Save the file and name it si.scf.in
- Open the command prompt on your PC and click (windows button + R and press ok) or search for command prompt.
- On the command prompt type “cd plus the destination folder of your quantum espresso. In my own case I input cd/”Program Files”/Quantum ESPRESSO 64-bit 5.3.0-mpich2/QE si . The “QE si” is the folder where my input file is. Press enter
- Then type pw. –in si.scf.in si.scf.out. Press enter. This will then run the program for you.
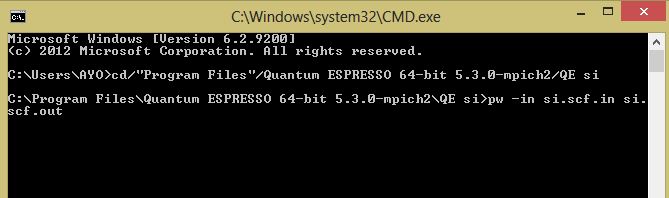
Once you see JOB DONE, then you have successfully installed quantum espresso on windows.
Note: I used windows 8 for the installation.
To download the silicon scf input file, click here